困扰数学家一个多世纪的难题,AI从生物学中找到线索
纽结理论长期困扰着数学家,但随着人工智能的进步,有研究者在生物学中找到了突破口,因为纽结结构存在于许多重要的生物分子中,例如蛋白质、DNA等。对于难以识别归类的复杂纽结结构,AI给出了令人惊讶的结果。
撰文 | Davide Michieletto(爱丁堡大学物理与天文学院)
编译 | 陈蕴灵
任何一个有经验的水手都明白,一个正确的绳结意味着生命的保障,无论是升起风帆还是固定锚链,绳子的作用取决于它的绳结。同样的道理也适用于维持人类生命体征的分子,例如最常见的蛋白质分子。
蛋白质是由氨基酸组成的大分子化合物,是构成生物体的基本物质之一。氨基酸通过肽键连接形成肽链,进而通过螺旋、折叠等方式形成复杂的空间结构,使分子能够相互耦合。长期以来,人们认为,尽管蛋白质可能高度缠绕,但在正常条件下它们不能形成纽结,因为这会阻碍蛋白质的折叠[译者注1]。
然而,在上世纪70年代,科学家发现了许多在拓扑意义上形成纽结结构的蛋白质(Knotted protein,也称打结蛋白),它们的天然结构呈现为开放的纽结(即,两端不连接)。尽管几何意义上,蛋白质(甚至DNA)可以看作一条(或几条)不闭合的曲线,但它们仍然能够形成纽结,从而影响其功能。事实上,在蛋白质数据库中,大约1%的蛋白质包含纽结结构。与绳子或者线不同,每一个纽结蛋白质都具有一个特有的纽结(见图1)。纽结蛋白质中最大的一类是SPOUT酶(它们组成了七类不同结构的甲基转移酶中的第二大类)。除了一种例外,其余所有SPOUT酶都形成了“三叶结”——三个环相互重叠的结构。
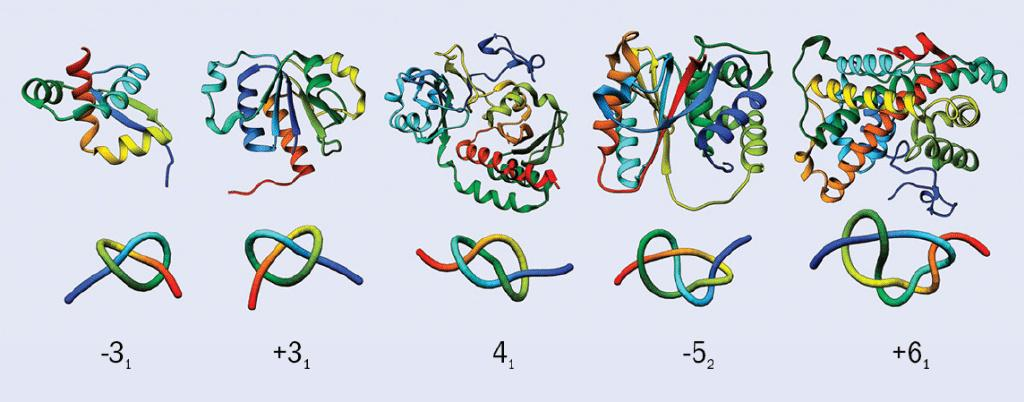

这一发现引发了一系列问题,比如:蛋白质分子的纽结结构是如何形成的,它们折叠的机制是什么,这些纽结又有什么作用,等等。有些证据表明,纽结蛋白质在极端温度下仍可以保持活性。但对于分子结构中纽结的普遍性及其对生物的影响,科学家们依然束手无策。当我们试图将对纽结的理解用于生物学和软物质问题上时,我们遇到了一个困扰科学家一个多世纪的数学问题。
曲折的历史:错误的涡旋原子理论
现代纽结理论的起源通常可以追溯到150多年前的一项著名实验——烟圈实验。1867年,彼得·格思里·泰特(Peter Guthrie Tait)邀请他的朋友——物理学家威廉·汤姆森(William Thomson,后来的开尔文勋爵),从格拉斯哥前往爱丁堡观看他演示制造成对的烟圈。令开尔文惊讶的是,这些烟圈异常稳定,能在房间里移动,甚至像橡胶一样相互弹开。烟圈事实上是一种“涡环”(vortex ring),其中气溶胶和颗粒物在同心小圆中旋转,这种运动赋予了烟圈稳定性。
当时,科学家们普遍认为宇宙中充满了一种被称为“以太”的物质,引力和电磁辐射以这种物质为媒介进行传播。开尔文推测,原子可能是以太中的稳定涡旋,类似于烟圈。他进一步认为,以太涡旋形成的纽结决定了不同的化学元素。泰特对开尔文的猜想产生了浓厚兴趣。在接下来的25年里,泰特与英国国教牧师托马斯·柯克曼(Thomas Kirkman)、美国数学家查尔斯·利特尔(Charles Little)以及詹姆斯·克拉克·麦克斯韦(James Clerk Maxwell)合作,制作了一张包含251个纽结的表格,其中最多有10个交叉点(见图2)。
随着科学家对原子结构的深入了解,涡旋原子模型逐渐被抛弃。1913年,亨利·莫塞莱(Henry Moseley)的研究最终推翻了这一理论,他证明化学元素的不同不是由它们的拓扑结构决定的,而是由原子核中质子数决定。尽管现在看来涡旋原子理论是错误的,但纽结理论仍作为数学的一个分支延续至今[译者注2]。

图 2:彼得·格思里·泰特和其他早期的纽结理论家花费多年时间编制了一份全面的纽结列表。上图摘自他们的结表,列出了最多9个交叉点的纽结——即“九阶纽结”[译者注3]。图片来源:University of St Andrews Library, ID sfQC3-T2-Vol-1-6.
识别纽结:寻找拓扑不变量
对于泰特及其同僚来说,为纽结分类是一项极为艰苦的工作。每当出现一个新的纽结时,他们都必须通过绘图和几何直观来检查其是否独特。泰特本人曾写道:“虽然我将许多形式截然不同但等价的结归为一组,但我不能绝对地确定这些分组本质上是否完全不同。”确实,1974年肯尼斯·佩尔科(Kenneth Perko)发现,泰特等人所列举的纽结表中有两个纽结实际上是等价的——它们现在被称为“佩尔科对”[2]。
如果想要更有说服力,我的学生乔尔杰·米哈伊洛维奇(Djordje Mihajlovic)开发了一款名为“识别纽结”(Spot a Knot)的在线游戏,这款游戏的目标是从图片中找出等价的纽结(见图3)。即便研究纽结多年,我自己还是经常出错。要想在纽结表中占有一席之地,一个纽结必须具有独特的拓扑结构,也就是说,除非将其拆开,否则它不能光滑形变成其他已知的结。拓扑学研究的是一个空间在光滑形变后的不变量。对拓扑学家而言,一个马克杯等价于一个甜甜圈,因为二者可以互相形变并且不会丢失内在的洞。但正如佩尔科对和米哈伊洛维奇的游戏所展示的那样,证明两个纽结不同并非易事。
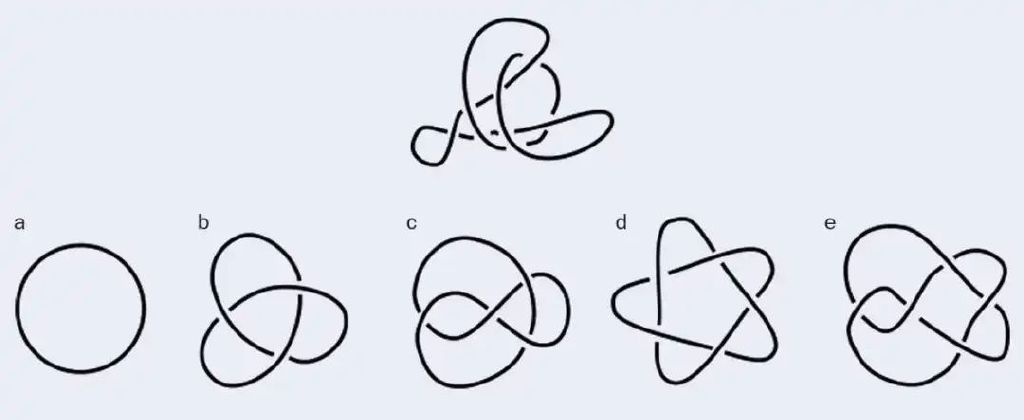
图 3:爱丁堡大学的博士生乔尔杰·米哈伊洛维奇开发了一款名为“识别纽结”的在线游戏。读者不妨尝试思考一下:在上图中,第一行的纽结等价于第二行的哪个呢?图片来源:Djordje Mihajlovic
在纽结理论中,用来刻画纽结性质的量称为“不变量”。纽结理论学家的终极梦想是找到一个不变量,能够根据拓扑结构用其对纽结进行分类(就像质子数一样,可以唯一确定一种化学元素)。这种“完全不变量”将赋予每一个纽结唯一的值,并且在光滑形变后保持不变。第一个纽结不变量是1928年由詹姆斯·亚历山大(James W. Alexander)提出的,称为亚历山大多项式(Alexander polynomial)。自那时起,数学家们提出了许多其他不变量,但对于每一个方法,人们都发现有无法进行唯一分类的案例。
一种构造拓扑不变量的方法是这样的:“沿着纽结走一遍,将每个交叉点编号为1, 2, 3, …, 2n(每个交叉点会遍历两次)。如果编号是偶数而且是从上方越过形成的交叉点,则将其符号标为负数(见图4)。最后,每个交叉点会被标记为一对整数,一个是偶数,一个是奇数。这一系列偶数就是该纽结的编码。”这个方法被称为Dowker–Thistlethwaite编码(简称DT编码),于1983年首次提出[3]。虽然DT编码可以分类许多简单的结,但与其他方法一样,它本身并不是拓扑不变量[译者注4]。
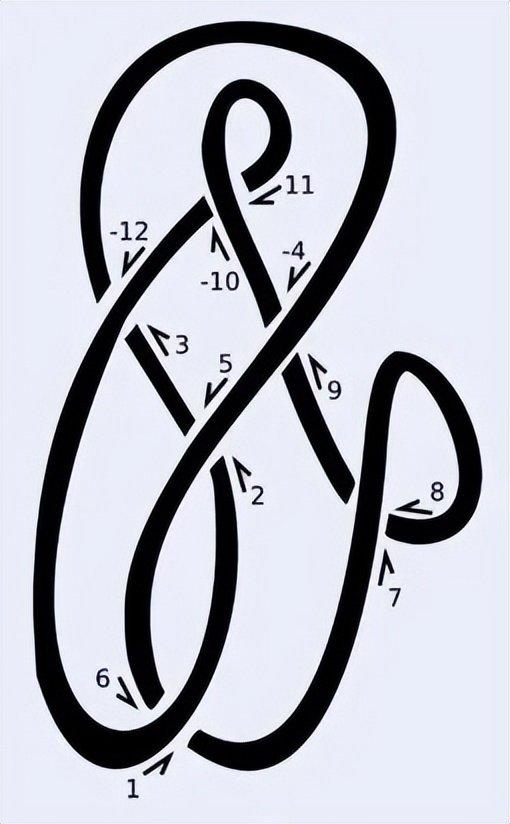
图 4:Dowker–Thistlethwaite记法是一种于1983年提出的纽结不变量。这种方法通过两次遍历纽结,为每个交叉点分配一个整数,最终的整数序列即表示为该纽结。图片来源:wikipedia
纽结不变量:代数与几何方法
亚历山大多项式属于所谓的“代数不变量”。它通过构造一个矩阵(其行数和列数与结的交叉点数量相同)并计算其行列式来确定。这类代数不变量是从结的二维投影中构造出来的,有点像纽结的影子,不过我们可以辨别每次交叉时哪个部分在上面。然而,像我这样的软物质物理学家希望对蛋白质和DNA等分子中的纽结进行分类,它们是三维的,并且不断受到热能的干扰,将这些分子简化为二维投影会抹去一些对其功能至关重要的空间特征。
一种有说服力的替代方法是使用“几何不变量”。这类不变量通过在三维空间中遍历纽结并通过计算某些几何属性(例如曲率)来确定。其中一种我非常喜欢的方式是计算绞拧数[译者注5](writhe,或称缠绕数),同样是由泰特提出的。绞拧数可以通过在二维投影中数“上方”和“下方”的交叉点,并用两者相减来计算(见图5b)。
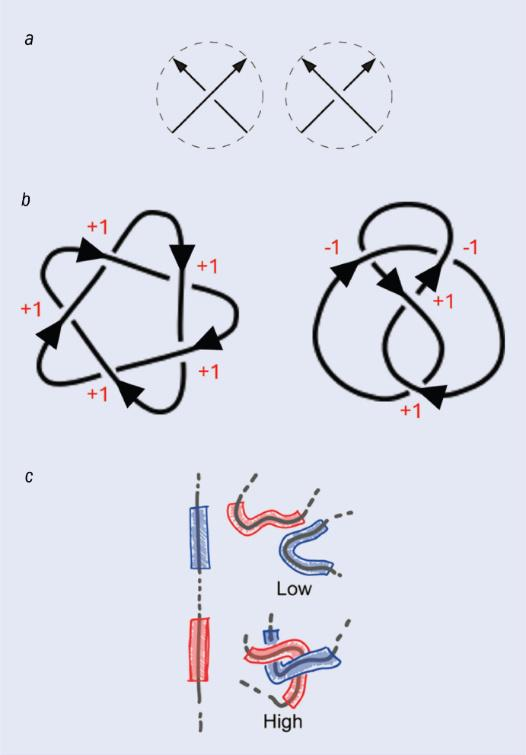
图 5:刻画纽结的一种方法是计算绞拧数,它量化了纽结的扭曲程度。(a) 每次纽结出现交叉时,交叉点可以被定义为上交叉(左图)或下交叉(右图)。绞拧数通过用上交叉点数量减去下交叉点数量来计算。(b) 绞拧数的计算示例:对于五叶结(左),它的绞拧数为+5;对于八字结(右),它的绞拧数为0。(c) 绞拧数还可以作为几何量在三维分子(如蛋白质)上进行计算。几何绞拧数可以作为整体量或局部量(在相邻的短链段之间)来计算。局部绞拧数值越大表明这些链段彼此缠绕得越紧。达维德·米基耶莱托及其同事证明,基于局部绞拧数训练的神经网络能够高精度地刻画纽结的拓扑结构。)图片来源:(a)参考文献[4]; (b) Djordje Mihajlovic; (c) 参考文献[5]
绞拧数不仅是代数量,也可以作为几何量来计算。想象在一个三维纽结(如蛋白质)上行走,每走一步计算你看到的交叉点的绞拧数。最后,将这些数值的平均值求出,就得到这个纽结的全局绞拧数。不幸的是,绞拧数并不是完全不变量。实际上,和代数不变量一样,没有任何几何不变量被证明能够对所有纽结进行唯一分类。
2021年,Google DeepMind的AI程序AlphaFold基本解决了困扰科学家几十年的一个问题——如何从氨基酸序列预测蛋白质的结构[6]。蛋白质的功能取决于其三维结构,因此AlphaFold成为研发药物和疾病研究的强大工具。
这使得我们不禁自问:人工智能是否能为纽结不变量问题提供类似的解决方案?
人工智能与绞拧数
一些研究者已经开始使用人工智能来探讨纽结的分类问题。最新的一些研究成果包括:2020年香港城市大学的Olafs Vandans团队[8]和2023年帕多瓦大学的Anna Braghetto团队的工作[9]。这些研究将不同的纽结视为一连串珠子,并通过给定的笛卡尔坐标、(后者中是)向量、珠子间的距离和角度等数据来训练神经网络,从而识别纽结的类型。这些研究取得了高精度的分类结果,但仅限于五个最简单的纽结。
我们希望将此方法推广到更复杂的拓扑结构,同时简化神经网络模型,并使用更小的训练数据集。为此,我们从大自然中获得灵感。在我们的身体中,DNA分子的纽结可以由所谓的“拓扑异构酶”(Topoisomerase)解开。尽管这些酶的体积只是DNA分子的千分之一,但它们可以切割并重新连接DNA链,从而有效地解开纽结。我们假设拓扑异构酶能够感知某种局部几何性质,使它们能够找到DNA分子中最紧密缠绕的部分。我们尝试使用密度、曲率等多种量来实现这一目标。最终,我们的结果又回到了起点——泰特和他的几何绞拧数。
除了计算整个纽结的绞拧数外,我们还可以计算局部绞拧数,这个量可以刻画某一链段的缠绕程度(见图5c)。我们发现计算局部绞拧数是一种定位长环状分子中纽结部分的有效方法[10]。基于这一结果,我们决定将局部绞拧数的数据提供给AI,这样它将有更大的胜算来识别复杂纽结。
这套理论成型后,我们开始构建神经网络进行测试。首先,我们通过模拟五个最简单的纽结的热运动生成训练数据集,提取数万个构型(见图6a)。随后,我们训练了两个神经网络:一个使用纽结的笛卡尔坐标,另一个使用局部绞拧数。在两种情况下,我们都对AI进行了监督,使用我们训练数据集的一个子集来告诉神经网络每个纽结的类型。我们要求神经网络对它们之前从未见过的简单结进行分类,以检验我们的方法。
当AI在简单的神经网络上使用笛卡尔坐标进行训练时,正确率只有80%,这与Vandans和Braghetto的成果类似。尽管这可能比我们大多数人在“识别纽结”游戏中的得分要高,但仍与我们的预期相差甚远。然而,当神经网络使用局部绞拧数进行训练时,结果令人震惊:它的正确率超过了99.9%。
更具挑战性的任务:局部绞拧数识别复杂纽结
尽管我对这一结果感到惊讶,但识别五个简单纽结的任务实际上相对平凡,我们完全可以通过现有的不变量完成(甚至极为敏锐的“识别纽结”玩家也可以完成)。因此,我们决定给神经网络一个更棘手的挑战。这一次,我们精心挑选三个纽结:康威(Conway)结、木下-寺坂纽结(Kinoshita–Terasaka,以下简称KT结),以及最简单的“平凡结”(unknot)。前两个纽结都有11个交叉点,并且是彼此的变形,因为它们除了在一个区域“翻转”外是完全相同的。它们共享许多的纽结不变量,同时也与平凡结共享一些不变量。我们发现,对于使用笛卡尔坐标训练的神经网络,康威结和KT结是无法区分的,但使用局部绞拧数训练的神经网络能够以99.9%的准确率识别它们。
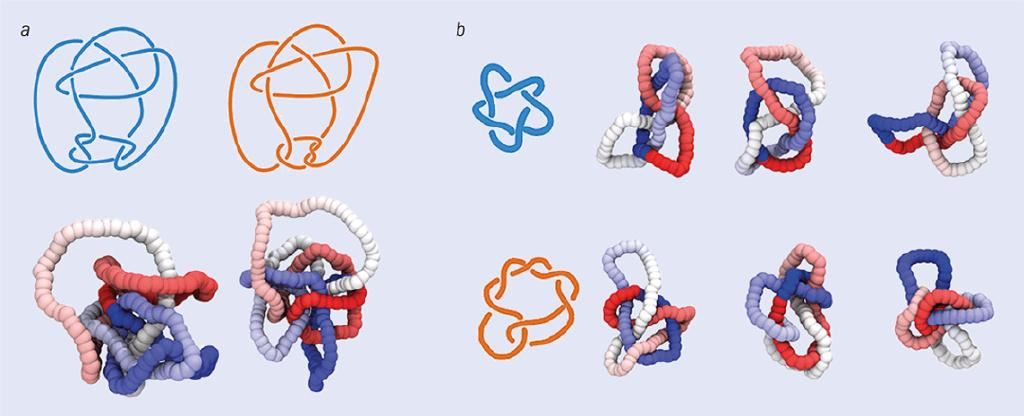
图 6:纽结的完全不变量在光滑形变下保持不变。不同的拓扑结构具有不同的完全不变量。(a)中的两幅图像展示的是同一个结吗?仅凭人类的直觉往往难以区分。事实上,这两幅图显示的是两种略微不同的结——康威结和KT结。由于很难区分这两个结,它们可以用来测试神经网络是否可以识别纽结。(b)展示了两种结的不同构型——51结(也称五叶结,上图)和72结(下图)。在达维德·米基耶莱托及其同事的神经网络研究中,五叶结是第一个训练数据集的一部分,而72结则包含在更大的数据集中。图片来源:(a)参考文献[5];(b) Davide Michieletto
我们的终极测试是将这种训练应用于更大范围的纽结。我们对250种结进行了模拟,这些结最多有10个交叉点(见图6b)。当神经网络使用笛卡尔坐标进行训练时,正确率只有20%。相比之下,我们使用局部绞拧数进行训练,AI可以在几秒内以95%的准确率分类所有250个结,这远超任何已知的算法或单一拓扑不变量[5]。
最后的验证——瓦西里耶夫不变量
在对纽结理论一无所知的情况下,神经网络通过自学,具备了一种长期跳脱于人类直觉之外的能力。实际上,人们仍在努力打开“黑箱”,理解神经网络究竟探索到了什么。我们发现,为了区分五个最简单的结,神经网络会将结上的每一对点的绞拧数相乘。值得注意的是,这个量等价于一个已存在的不变量,称为“二阶瓦西里耶夫(Vassiliev)不变量”。瓦西里耶夫不变量是通过将局部绞拧数矩阵的二元组、三元组、四元组……直到n元组相乘来计算的。顺便提一下,二阶瓦西里耶夫不变量也是康威多项式的二次项系数,这是一种我们之前提到的代数不变量。一个仍未被验证的猜想认为,能够通过积分计算的瓦西里耶夫不变量完备集可能就是长期寻求的完全不变量。
我们很高兴地发现,神经网络能够通过计算更高阶的瓦西里耶夫不变量来适应更复杂的结。例如,为了唯一分类五个简单结,神经网络仅需计算二阶瓦西里耶夫不变量。但在250个结的数据集中,它可能会计算三阶或四阶的瓦西里耶夫不变量。
几何不变量和代数不变量是通过非常不同的数学方法计算的。AI能够发现它们之间的联系非常令人兴奋,这也使我们更接近完全不变量。
纽结的确很重要
在短短三年内,AlphaFold生成了数百万种蛋白质,其中大部分尚未被完全研究。2023年,由华沙大学的乔安娜·苏尔科夫斯卡(Joanna Sulkowska)领导的一组研究人员预测,通过AlphaFold生成的蛋白质中有多达2%是纽结蛋白质,其中最复杂的结有六个交叉点[11]。2022年,约翰内斯·古滕贝格大学的彼得·维尔瑙(Peter Virnau)在AlphaFold2数据集中发现了一种具有七个交叉点的纽结蛋白质[12]。这种蛋白质从未在实验中观察到,因此我们不无理由相信可能还有更复杂的纽结蛋白质存在。
纽结不仅存在于生物学中。有研究发现,纽结的拓扑结构会影响冰和水凝胶的热力学和材料属性,这意味着未来我们可能会利用拓扑性质设计新材料。我们需要更有效的方法来识别分子和材料中的纽结结构,希望我们的发现能为这一探索提供指导。
在2004年,加拿大的三位研究人员利用他们大学的计算集群将泰特最初编制的结表扩展至19个交叉点的交错纽结,构造出超过六十亿种独特的纽结结构[7]。泰特花了25年创建他自己的纽结列表,他可能会惊讶于一百年后,一台机器竟然能在短短几天内将他的工作扩展超过五个数量级。纽结理论中目前最大的未解之谜仍然是寻找完全不变量。现在,既然有了AI的助力,下一步的进展可能让我们感到惊讶。
译者注
[1]蛋白质可凭借相互作用在特定环境下自我组装,这个过程被称为蛋白质折叠。蛋白质折叠问题是分子生物学领域尚未解决的一个重大课题。
[2]数学上,我们定义一个纽结为空间中的一个闭合曲线,并且该曲线在任何地方不会出现自相交。
[3]此处原文The above image is extracted from their table of knots up to seven crossings – “the first seven orders of knottiness”,表述有误。从图中不难看出,泰特等人列出的结表中包含了最多9个交叉点。图2实际上列出了非平凡纽结的前七种情况,即交叉点的个数大于等于3且小于等于9。(注意,交叉点小于等于2的纽结可以光滑形变为平凡结。)
[4]本段原文有矛盾,“A recipe for such a topological invariant could be something like this……”,“it isn’t a complete invariant”。DT编码只是用来构造拓扑不变量的一种方法,它本身并不是一个不变量。DT编码提供了一个直接的符号化表示,使得纽结可以转换成一串数字,便于分析和计算。人们可以进一步推导出亚历山大多项式等重要的纽结不变量。另外,使用DT编码,目前还无法构造出真正的完全不变量。
[5] 此处原文有误,“One such invariant that I am fond of is the ‘writhe’”。和DT编码一样,绞拧数本身并不是拓扑不变量,但可以用来构造拓扑不变量。不同的是,绞拧数用于描述纽结二维投影图的几何特性(而非单一的代数性质,具体见下文)。例如绞拧数可以用来构造纽结的自交数,在区分镜像纽结、描述纽结几何特性和某些物理系统(如DNA链)的研究中有重要应用。同样,使用绞拧数,目前还无法构造出真正的完全不变量。
参考文献
[1]Dabrowski-Tumanski, Pawel, and Joanna I. Sulkowska. "To tie or not to tie? That is the question." Polymers 9.9 (2017): 454.
[2]Perko, Kenneth A. "On the classification of knots." Proceedings of the American Mathematical Society 45.2 (1974): 262-266.
[3]Dowker, Clifford H., and Morwen B. Thistlethwaite. "Classification of knot projections." Topology and its Applications 16.1 (1983): 19-31.
[4]Ippoliti, Emiliano. "On the heuristic power of mathematical representations." Synthese 200.5 (2022): 407.
[5]Sleiman, Joseph Lahoud, et al. "Geometric learning of knot topology." Soft Matter 20.1 (2024): 71-78.
[6]Jumper, John, et al. "Highly accurate protein structure prediction with AlphaFold." nature 596.7873 (2021): 583-589.
[7]Vandans, Olafs, et al. "Identifying knot types of polymer conformations by machine learning." Physical Review E 101.2 (2020): 022502.
[8]Braghetto, Anna, et al. "Machine learning understands knotted polymers." Macromolecules 56.7 (2023): 2899-2909.
[9]Sleiman, Joseph L., et al. "Geometric Predictors of Knotted and Linked Arcs." ACS Polymers Au 2.5 (2022): 341-350.
[10]Perlinska, Agata P., et al. "AlphaFold predicts novel human proteins with knots." Protein Science 32.5 (2023): e4631.
[11]Brems, Maarten A., et al. "AlphaFold predicts the most complex protein knot and composite protein knots." Protein Science 31.8 (2022): e4380.
[12]Rankin, Flint, Schermann. "Enumerating the prime alternating knots, Part I." Journal of Knot Theory and Its Ramifications 13.01 (2004): 57-100.
本文基于知识共享许可协议(CC BY-NC)译自Davide Michieletto, Spot the knot: using AI to untangle the topology of molecules.
